

Shopping Cart
Remove All Your shopping cart is currently empty
Your shopping cart is currently empty
TargetMol Star Molecule—Mito-TEMPO (Cat. No. T19428, CAS 1091902-09-5), A Mitochondria-Targeted SOD Mimetic for ROS, Oxidative Stress, and Mitochondrial Dysfunction Research
Background
Mito-TEMPO (T19428) is a mitochondria-targeted superoxide dismutase (SOD) mimetic that plays a critical role in modulating mitochondrial reactive oxygen species (ROS) dynamics. By selectively accumulating within mitochondria, Mito-TEMPO scavenges superoxide anions and alkyl radicals, thereby mitigating oxidative stress at its primary intracellular source. This targeted antioxidant activity prevents mitochondrial oxidation, which is a pivotal event in the initiation of necrosis and apoptosis. The compound’s mechanism of action centers on mimicking endogenous SOD enzymes, catalyzing the dismutation of superoxide radicals into less reactive species, thus preserving mitochondrial integrity and function. Within the broader context of mitochondrial metabolism, ROS serve as signaling molecules that regulate cellular homeostasis but, when in excess, contribute to oxidative damage and cell death pathways. Mito-TEMPO dynamically interacts with this pathway by attenuating the overproduction of mitochondrial ROS, thereby modulating redox-sensitive signaling cascades and preventing downstream pathological consequences. In research settings, Mito-TEMPO is widely employed to dissect the role of mitochondrial oxidative stress in various cellular models, including studies on aging, neurodegeneration, ischemia-reperfusion injury, and metabolic disorders. Its utility extends to probing the mechanistic underpinnings of ROS-mediated mitochondrial dysfunction and evaluating potential interventions that target mitochondrial redox balance. By providing a precise tool to modulate mitochondrial ROS, Mito-TEMPO facilitates a deeper understanding of mitochondrial metabolism and its impact on cell fate decisions without directly altering other cellular antioxidant systems. This specificity makes it invaluable for elucidating the complex interplay between mitochondrial ROS production and cellular signaling pathways involved in oxidative stress responses [1,2].
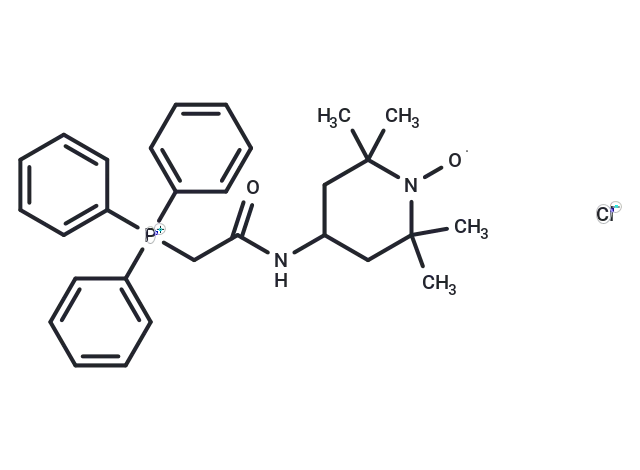
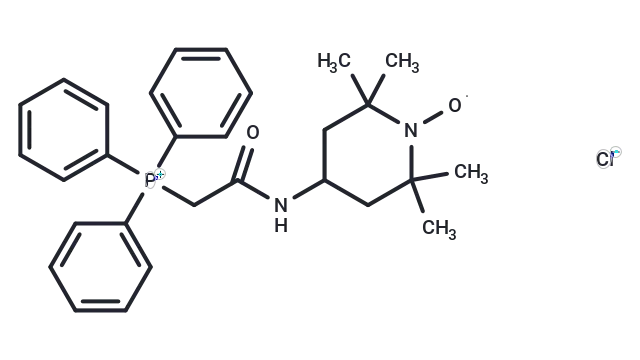
Molecular Structure of Mito-TEMPO
Literature review
2.1 Chlamydia pneumoniae Infection Induces Vascular Smooth Muscle Cell Migration and Atherosclerosis Through Mitochondrial Reactive Oxygen Species-Mediated JunB-Fra-1 Activation
In this study, Mito-TEMPO(T19428) was shown to inhibit the mitochondrial reactive oxygen species (mtROS) generated by Chlamydia pneumoniae infection in vascular smooth muscle cells (VSMCs). Pretreatment with Mito-TEMPO eliminated the increase in mtROS levels induced by C. pneumoniae and significantly reduced VSMC migration, as demonstrated by both Transwell and wound healing assays. Furthermore, the expression of proteins Fra-1, JunB, and MMP2—upregulated during C. pneumoniae infection and associated with VSMC migration—were all suppressed by Mito-TEMPO. These experimental results collectively indicate that Mito-TEMPO suppresses key molecular and cellular processes triggered by mitochondrial ROS production in response to infection, leading to reduced VSMC migration. The figures associated with these findings are Fig.3, which shows the inhibition of VSMC migration, and Fig.6, where the suppression of Fra-1 and MMP2 expression by Mito-TEMPO is confirmed. Overall, the drug inhibited mitochondrial ROS and downstream factors, thereby decreasing cell migration related to infection.[3]
2.2 Hypoxia-preconditioned bone marrow–derived mesenchymal stem cells protect neurons from cardiac arrest–induced pyroptosis
In this study, Mito-TEMPO(T19428), a mitochondrial-targeted ROS scavenger, was applied in a co-culture system with hypoxia preconditioned bone marrow mesenchymal stem cells (HP-BMSCs) following oxygen-glucose deprivation (OGD). The addition of Mito-TEMPO further improved mitochondrial membrane potential recovery in neurons beyond the effect of HP-BMSCs alone, indicating enhanced mitochondrial function. Mito-TEMPO also further reduced both intracellular and mitochondrial reactive oxygen species (ROS) levels in neurons when combined with HP-BMSCs. Furthermore, the presence of Mito-TEMPO led to a greater decrease in the expression of neuronal mitochondrial damage markers such as FIS1, COX IV, and TOM20 compared to HP-BMSCs without Mito-TEMPO. These results explicitly indicate that Mito-TEMPO effectively alleviated mitochondrial oxidative stress and enhanced mitochondrial protection in neurons exposed to OGD in the experimental model. Thus, in this study, Mito-TEMPO worked synergistically with HP-BMSCs to reduce oxidative damage in neuronal mitochondria.[4]
2.3 Elevation of ISG15 promotes diabetic kidney disease by modulating renal tubular epithelial cell pyroptosis
In this study, Mito-TEMPO(T19428) functioned as a mitochondrial reactive oxygen species (mtROS) scavenger that reduced the overproduction of mtROS caused by ISG15 overexpression in tubular epithelial cells (TECs). The application of Mito-TEMPO alleviated the excessive mtROS generation induced by ISG15, demonstrating a direct effect on mitochondrial oxidative stress. Additionally, Mito-TEMPO reversed the up-regulation of pyroptosis-associated proteins including NLRP3, GSDMD, GSDMD-N, as well as the injury marker KIM1, all of which were elevated following ISG15 overexpression. These findings indicate that Mito-TEMPO mitigated ISG15-mediated mitochondrial injury and associated pyroptotic signaling events by reducing mitochondrial oxidative stress. Therefore, Mito-TEMPO exerted a protective effect in the study by reducing mitochondrial reactive oxygen species and downstream pyroptosis-related protein expression in HG-induced TEC injury models.[5]
2.4 Cardiomyocyte-specific Piezo1 deficiency mitigates ischemia-reperfusion injury by preserving mitochondrial homeostasis
Mito-TEMPO(T19428) demonstrated significant effects in this study by suppressing ischemia/reperfusion (I/R)-induced oxidative stress and inflammation. Specifically, Mito-TEMPO treatment effectively reduced reactive oxygen species (ROS) levels generated by I/R, subsequently inhibiting leukocyte infiltration as shown by reduced macrophage and neutrophil presence. The drug also attenuated the expression of inflammatory cytokines related to both I/R in vivo and hypoxia/reoxygenation (H/R) in vitro conditions. Furthermore, Mito-TEMPO diminished accumulation of mitochondrial and cellular ROS induced by H/R in cardiomyocytes. Experimentally, this drug reduced myocardial infarction size, lowered serum lactate dehydrogenase (LDH) levels, and decreased apoptosis in cardiomyocytes and neonatal mouse cardiomyocytes (NMCMs). It also enhanced cell viability and lowered LDH release in NMCM cultures. These findings collectively indicate that Mito-TEMPO(T19428) exerts a protective effect in the context of myocardial ischemia-reperfusion injury through suppression of oxidative stress and the associated inflammatory response.[6]
2.5 The role of mitochondrial reactive oxygen species in initiating mitochondrial damage and inflammation in wasp-venom-induced acute kidney injury
Mito-TEMPO(T19428) acted as a mitochondrial reactive oxygen species (mtROS) scavenger to inhibit the mitochondrial ROS burst in the kidney in a mouse model of wasp-venom-induced acute kidney injury. Administration of Mito-TEMPO led to the downregulation of mtROS levels, which reversed renal damage and mitochondrial dysfunction induced by wasp venom. Furthermore, Mito-TEMPO decreased expression of the stimulator of interferon gene (STING) and reduced inflammation in the kidney tissue. The drug also alleviated decreases in mitochondrial transcription factor A (TFAM) expression and mitochondrial DNA copy numbers observed in the kidney tissue of model mice. Overall, Mito-TEMPO(T19428) showed protective effects on mitochondrial integrity and kidney inflammation by suppressing mtROS in this specific experimental setup.[7]
Reference
[1] 1. Dikalova AE, et al. Mitochondria-targeted antioxidant Mito-TEMPO decreases diabetic cardiomyopathy. Free Radic Biol Med. 2010;48(11):1483-1491.
[2] 2. Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. 2008;1777(7-8):1028-1031.
[3] Zhao X, Miao G, Zhang L, Zhang Y, Zhao H, Xu Z, et al.. Chlamydia pneumoniae Infection Induces Vascular Smooth Muscle Cell Migration and Atherosclerosis Through Mitochondrial Reactive Oxygen Species-Mediated JunB-Fra-1 Activation. Frontiers in Cell and Developmental Biology. 2022;10():.
[4] Tang X, Zheng N, Lin Q, You Y, Gong Z, Zhuang Y, et al.. Hypoxia-preconditioned bone marrow–derived mesenchymal stem cells protect neurons from cardiac arrest–induced pyroptosis. Neural Regeneration Research. 2024;20(4):1103-1123.
[5] Huang L, Chen X, Shao Y, Deng S, Wang C, Chen J, et al.. Elevation of ISG15 promotes diabetic kidney disease by modulating renal tubular epithelial cell pyroptosis. Clinical and Translational Medicine. 2025;15(6):.
[6] Xu H, Chen X, Luo S, Jiang J, Pan X, He Y, et al.. Cardiomyocyte-specific Piezo1 deficiency mitigates ischemia-reperfusion injury by preserving mitochondrial homeostasis. Redox Biology. 2025;79():103471.
[7] Xia L, Yuan H, Gao Z, Lv Y, Xu L, Hu F. The role of mitochondrial reactive oxygen species in initiating mitochondrial damage and inflammation in wasp-venom-induced acute kidney injury. Journal of Toxicologic Pathology. 2025;38(1):17-26.
An essential round-up of science news, opinion and analysis, delivered to your inbox every weekday.
 Hello! How can I help you today?
Hello! How can I help you today? Copyright © 2015-2026 TargetMol Chemicals Inc. All Rights Reserved.