
Shopping Cart
Remove All Your shopping cart is currently empty
Your shopping cart is currently empty
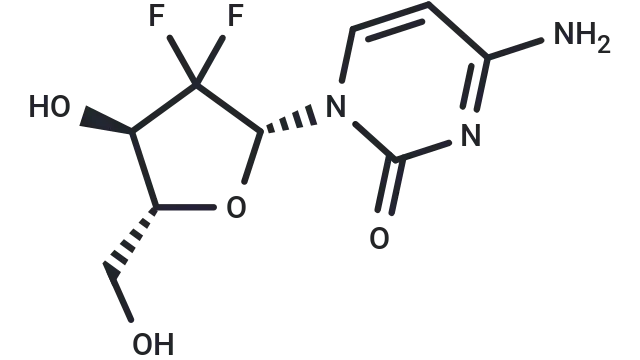
TargetMol Star Molecule—Gemcitabine (Cat. No. T0251, CAS 95058-81-4), A Nucleoside Analog Driving DNA Replication Stress, Autophagy, and Apoptosis
Background
Gemcitabine (T0251) is a synthetic cytosine nucleoside analog that functions primarily as an inhibitor of DNA synthesis, exerting its biological effects through incorporation into DNA and subsequent disruption of nucleic acid metabolism. As a nucleoside antimetabolite, Gemcitabine is phosphorylated intracellularly to its active diphosphate and triphosphate forms, which competitively inhibit ribonucleotide reductase and become incorporated into DNA strands, respectively. This dual mechanism results in the depletion of deoxyribonucleotide pools and premature termination of DNA chain elongation, effectively halting DNA replication. The interruption of DNA synthesis triggers cellular stress responses, including the activation of programmed cell death pathways such as apoptosis, as well as the induction of autophagy, a catabolic process that degrades damaged organelles and proteins to maintain cellular homeostasis.
The signaling pathways modulated by Gemcitabine encompass key components of DNA damage response and cell fate determination. By inducing DNA replication stress, Gemcitabine activates checkpoint kinases and p53-dependent pathways that orchestrate apoptosis. Concurrently, autophagy is stimulated as a cytoprotective mechanism, involving autophagosome formation regulated by ATG proteins and LC3 lipidation. The dynamic interplay between apoptosis and autophagy under Gemcitabine exposure reflects a complex cellular attempt to balance survival and death signals in response to nucleoside analog-induced genotoxic stress.
In research contexts, Gemcitabine is extensively utilized as a molecular tool to investigate mechanisms of DNA synthesis inhibition, nucleoside metabolism, and the crosstalk between autophagy and apoptosis. Its ability to induce DNA damage and modulate cell death pathways makes it valuable for studying cellular responses to genotoxic agents, elucidating the regulation of autophagy-related genes, and exploring the molecular determinants of cell cycle arrest. Furthermore, Gemcitabine serves as a probe to dissect the roles of ribonucleotide reductase and DNA polymerases in maintaining genomic integrity. The compound’s utility extends to modeling tumor cell biology in vitro, enabling researchers to analyze the cellular consequences of impaired DNA replication and the subsequent activation of stress-induced signaling cascades.
Overall, Gemcitabine (T0251) represents a critical biochemical tool for dissecting the molecular underpinnings of nucleoside analog-induced cytotoxicity, providing insights into the integration of DNA synthesis inhibition with autophagic and apoptotic pathways. Its application in experimental systems continues to advance understanding of cellular homeostasis mechanisms under conditions of metabolic and genotoxic stress, thereby informing broader biological research on cell cycle regulation and programmed cell death [1,2].
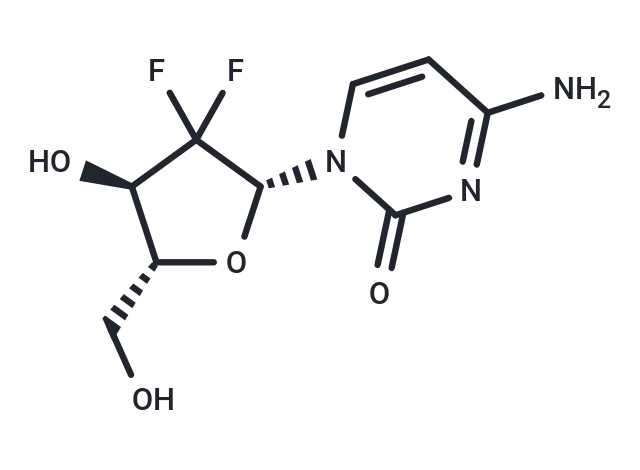
Molecular Structure of Gemcitabine
Literature review
2.1 A serum-derived 3D tumor model platform for personalized prediction and monitoring of chemotherapeutic response in pancreatic ductal adenocarcinoma
Gemcitabine(T0251) demonstrated cytotoxic effects in pancreatic ductal adenocarcinoma (PDAC) patient serum-derived educated spheroids by significantly decreasing cell viability to or below the target-independent cell killing (TICK) value, confirming its true therapeutic efficacy in vitro. Experimental results from four metastatic PDAC patients revealed differential sensitivity to gemcitabine monotherapy: one patient showed no response, two patients displayed sensitivity to gemcitabine alone and the Gem-Pac combination, while one patient responded only to gemcitabine monotherapy but not the combination. Additionally, spheroids derived from seven PDAC patients treated with gemcitabine confirmed reduced viability consistent with the clinical treatment protocol. Notably, the predictive accuracy of gemcitabine response was dependent on the timing of blood sample collection; serum samples taken within one month before or after therapy initiation correlated fully with clinical outcomes, whereas specimens collected more than one month prior to treatment displayed discordant in vitro responses. These findings illustrate the cytotoxic effect of gemcitabine in this patient-derived spheroid model and highlight the importance of precise timing for sample collection to ensure accurate prediction and monitoring of drug response.[3]
2.2 Lycorine hydrochloride inhibits cholangiocarcinoma through cholesterol biosynthesis and PTPN11 nuclear translocation
Gemcitabine(T0251) was evaluated in combination with Lycorine Hydrochloride (LY) in intrahepatic cholangiocarcinoma (ICC) cell models, where the combined use exhibited a potent synergistic anti-tumor effect across various tumor types. The combination treatment significantly reduced RBE cell viability beyond the effects of either agent alone. The IC50 of Gemcitabine in RBE cells was established at 5 µM, and combining concentrations below the IC50 values of both drugs still achieved enhanced efficacy. Mechanistically, the LY and Gemcitabine combination effectively downregulated the expressions of key enzymes in cholesterol biosynthesis, FDFT1 and SQLE, which were previously identified as oncogenic factors in ICC. This suggests that the synergy of Gemcitabine with LY may be mediated, at least in part, through modulation of cholesterol biosynthesis and associated signaling pathways. The study’s data demonstrate that Gemcitabine combined with LY possesses superior anti-tumor activity by leveraging interactions affecting tumor cell viability and molecular targets linked to tumor progression.[4]
2.3 Niraparib perturbs autophagosome-lysosome fusion in pancreatic ductal adenocarcinoma and exhibits anticancer potential against gemcitabine-resistant PDAC
Gemcitabine(T0251) serves as the cornerstone drug for pancreatic ductal adenocarcinoma (PDAC) treatment; however, resistance to gemcitabine remains a significant challenge. The study reports that autophagy activation and strengthened DNA damage repair are important factors contributing to gemcitabine resistance. Experimental results indicated that gemcitabine-resistant PDAC cells display increased sensitivity to the PARP inhibitor niraparib relative to wild type cells. Notably, combining gemcitabine with niraparib produced a significant antagonistic effect in wild type PDAC cell lines, but this antagonism was abolished in gemcitabine-resistant PDAC cell lines. This suggests the interaction between gemcitabine and niraparib depends on the resistance status of the cells. These findings highlight the mechanistic role of autophagy and DNA repair processes in modulating gemcitabine resistance and suggest different cellular responses depending on resistance profiles.[5]
Reference
[1] 1. Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22(4 Suppl 11):3-10.
[2] 2. Chen Z, Wang Y, Zhang X, et al. Gemcitabine induces autophagy and apoptosis through the AMPK/mTOR signaling pathway in pancreatic cancer cells. Cancer Lett. 2018;438:1-11.
[3] Cherradi S, Roux S, Dupuy M, Assenat E, Duong H. A serum-derived 3D tumor model platform for personalized prediction and monitoring of chemotherapeutic response in pancreatic ductal adenocarcinoma. Translational Oncology. 2026;64():102659.
[4] Zhao F, Peng S, Zou L, Zhong M, Huang Y, Wang P, et al.. Lycorine hydrochloride inhibits cholangiocarcinoma through cholesterol biosynthesis and PTPN11 nuclear translocation. Cell Communication and Signaling. 2025;23(1):.
[5] Yao Z, Zhang H, Huang K, Huang G, Xi P, Jiang L, et al.. Niraparib perturbs autophagosome-lysosome fusion in pancreatic ductal adenocarcinoma and exhibits anticancer potential against gemcitabine-resistant PDAC. Translational Oncology. 2025;51():102206.

An essential round-up of science news, opinion and analysis, delivered to your inbox every weekday.
 Hello! How can I help you today?
Hello! How can I help you today? Copyright © 2015-2026 TargetMol Chemicals Inc. All Rights Reserved.