
Shopping Cart
Remove All Your shopping cart is currently empty
Your shopping cart is currently empty
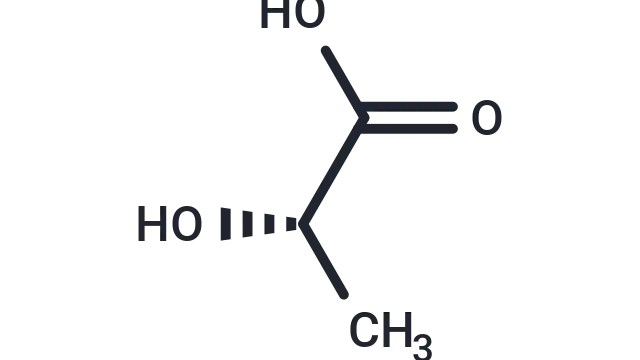
TargetMol Star Molecule—Mdivi-1 ( T1907, CAS 338967-87-6), the “brake” for mitochondrial dynamics research, a classic permeable small-molecule inhibitor
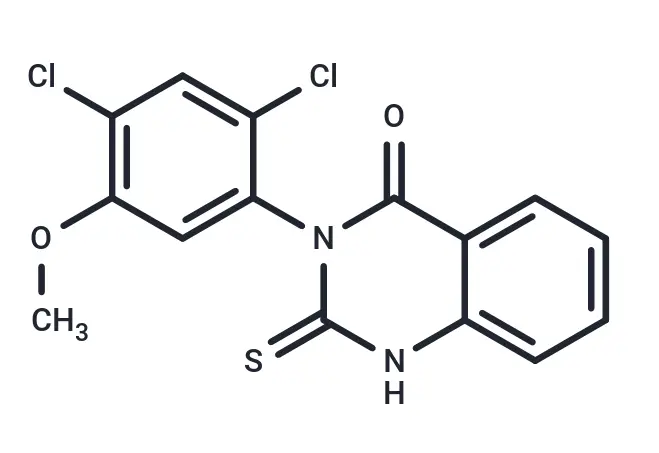
Molecular Structure of Mdivi-1
1. Background
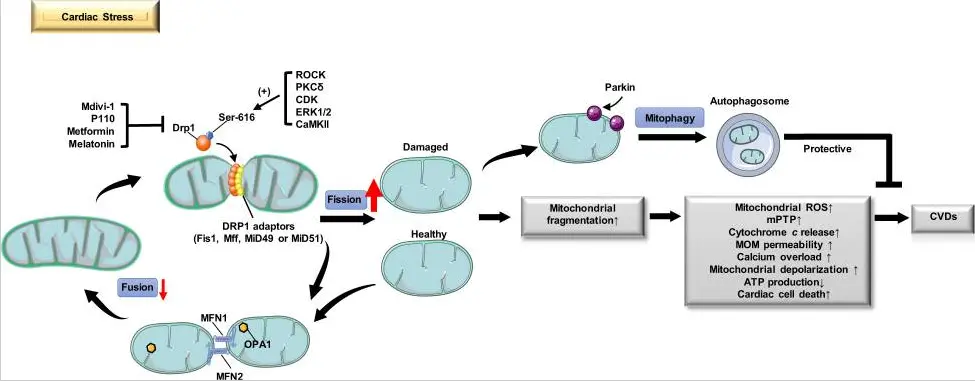
Mitochondrial fission inhibitors are a class of small-molecule compounds or peptides capable of suppressing excessive mitochondrial fission and maintaining mitochondrial morphological and functional homeostasis; their primary target is the mitochondrial fission core protein Drp1. These drugs do not completely block normal physiological mitochondrial fission; rather, they exert a cytoprotective effect by inhibiting abnormally activated fission pathways and correcting mitochondrial kinetic imbalances in pathological conditions.
Mitochondrial fission inhibitors primarily block the fission process through multiple mechanisms. Most drugs directly inhibit the GTPase activity of Drp1, preventing it from hydrolyzing GTP to generate conformational changes and contractile force. Some inhibitors interfere with the translocation and assembly of Drp1 from the cytoplasm to the outer mitochondrial membrane, or block the binding of Drp1 to membrane receptors such as Mff, MiD49/51, and Fis1, thereby preventing the formation of constriction loops. Through these mechanisms, inhibitors can effectively reduce mitochondrial fragmentation, stabilize the mitochondrial membrane potential, decrease reactive oxygen species (ROS) production, and reduce the release of apoptosis-related factors.
The applications of mitochondrial fission inhibitors are primarily focused on disease models and pathological processes involving excessive mitochondrial fission. In neurological disorders, they are commonly used in neuroprotective research for conditions such as cerebral ischemia-reperfusion injury, Alzheimer’s disease, and Parkinson’s disease, where they can mitigate neuronal death and functional impairment. In the cardiovascular field, these drugs can improve myocardial ischemia-reperfusion injury, reduce myocardial cell apoptosis, and minimize the area of myocardial infarction. Furthermore, in cancer research, mitochondrial fission inhibitors can alter the metabolic characteristics of tumor cells and enhance sensitivity to chemotherapy drugs. In conditions such as inflammation, organ injury, and certain metabolic diseases, they can also exert a protective effect by restoring mitochondrial homeostasis.
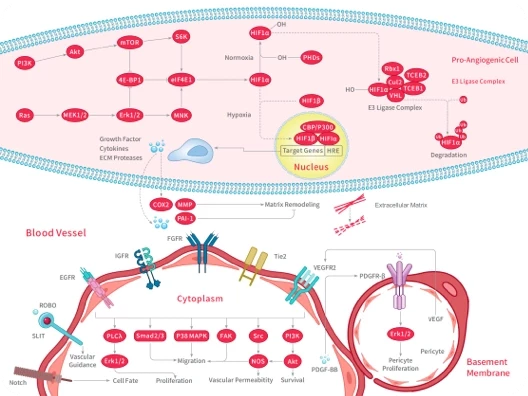
Mitochondrial Fission and Cardiovascular Disease [1]
The application of Mdivi-1 focuses on pathological conditions associated with excessive mitochondrial fragmentation, playing a significant role in various disease research models. In the field of neurological disorders, it is widely used in models of cerebral ischemia/reperfusion injury, Alzheimer’s disease, Parkinson’s disease, and epilepsy, effectively reducing neuronal death, inhibiting mitochondrial fragmentation, and stabilizing mitochondrial membrane potential, thereby improving neurological function and cognitive impairment.
In cardiovascular diseases, Mdivi-1 can improve myocardial ischemia/reperfusion injury, myocardial infarction, obesity-related cardiomyopathy, and traumatic hemorrhagic shock. It reduces myocardial cell apoptosis, aids in the restoration of mitochondrial structure and respiratory function, and extends the survival time of experimental animals. In cancer research, it inhibits Drp1 activity in tumor cells, blocks metabolic reprogramming and migration capabilities, and simultaneously enhances sensitivity to chemotherapy drugs, providing important experimental evidence for tumor therapy research targeting mitochondrial metabolism.
Furthermore, in other pathological models such as retinal ischemia, subarachnoid hemorrhage, and inflammation-related organ injury, Mdivi-1 can also mitigate tissue damage by inhibiting excessive mitochondrial fission, offering a potential strategy for clinical translational research on these diseases.
2. Selected Literature
2.1 Article Titles:Inhibition of DRP1-dependent mitochondrial fission by Mdivi-1alleviates atherosclerosis through the modulation of M1polarization

Study Overview: This study aims to investigate the effects and mechanisms of Mdivi-1, a Drp1-dependent mitochondrial fission inhibitor, on atherosclerosis (AS). The study confirmed that in high-fat diet-induced ApoE⁻/⁻ mice and ox-LDL-stimulated macrophages, excessive Drp1 activation triggers excessive mitochondrial fission and the accumulation of mitochondrial reactive oxygen species (mito-ROS), which in turn activates the NLRP3 inflammasome. This promotes the polarization of macrophages toward the pro-inflammatory M1 phenotype and the formation of foam cells, ultimately accelerating the progression of atherosclerotic plaques. Treatment with Mdivi-1 inhibits Drp1 (Ser616) phosphorylation and blocks excessive mitochondrial fission, thereby reducing M1 polarization and inflammatory responses via the mito-ROS/NLRP3 pathway, which alleviates atherosclerotic lesions. This suggests that Drp1-dependent mitochondrial fission may serve as a potential therapeutic target for atherosclerosis. [2]
This study utilized ApoE⁻/⁻ mice and RAW264.7 macrophages to investigate the effects and mechanisms by which Mdivi-1 improves atherosclerosis. Animal experiments included groups fed a normal diet, a high-fat diet, and a high-fat diet supplemented with different doses of Mdivi-1, with a 12-week intervention period. Cell experiments used ox-LDL induction, followed by treatment with Mdivi-1, MCC950, Mito-TEMPO, or Drp1 knockdown. Oil Red O staining, blood lipid levels, inflammatory factors, mitochondrial function, and protein expression were assessed. Results showed that Mdivi-1 reduced plaque area, inhibited M1 polarization and NLRP3 activation, and decreased Drp1 phosphorylation, mitochondrial fission, and mito-ROS production; Drp1 knockdown mimicked these effects. Mdivi-1 reduces M1 polarization and foam cell formation via the mito-ROS/NLRP3 pathway, thereby alleviating atherosclerosis.
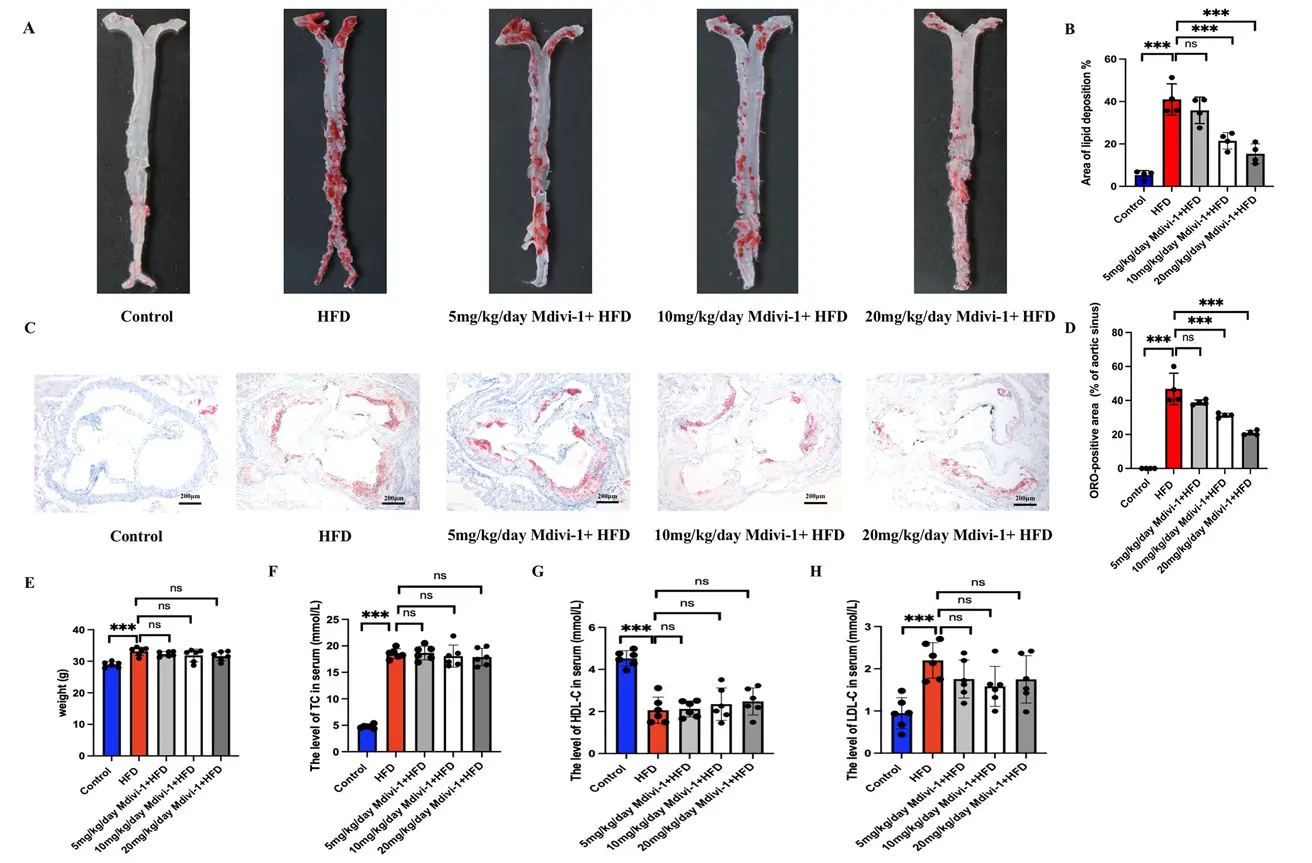
Mdivi-1 Reduces Atherosclerotic Plaques in ApoE⁻/⁻ Mice
2.2 Article Title:Lactate facilitated mitochondrial fission-derived ROS to promote pulmonary fibrosis via ERK/DRP-1signaling

Study Overview: This study reveals that lactate induces excessive mitochondrial fission via the ERK/DRP1 signaling axis, leading to the production of large amounts of mitochondrial reactive oxygen species (mtROS). This, in turn, activates NF-κB/P65 nuclear translocation, promotes pulmonary fibroblast activation and collagen deposition, and ultimately exacerbates bleomycin (BLM)-induced pulmonary fibrosis. The use of an ERK inhibitor (Ulixertinib), a DRP1 inhibitor (Mdivi-1), or an mtROS scavenger (Mito-TEMPO) can all block this pathway and improve pulmonary fibrosis, providing new therapeutic targets for idiopathic pulmonary fibrosis (IPF). [3]
This study established a mouse model of pulmonary fibrosis using bleomycin (BLM), with control, model, and intervention groups treated with sodium oxalate, ulixertinib, Mdivi-1, and Mito-TEMPO. In vitro experiments utilized MRC5 human lung fibroblasts and primary lung fibroblasts, which were induced with lactate and TGF-β and treated with the respective inhibitors. Mechanisms were validated through histochemical staining, biochemical assays, qPCR, Western blotting (WB), mitochondrial fluorescence staining, and cellular functional assays. Results showed that BLM elevated lactate and ROS levels; lactate induced mitochondrial fission and mtROS accumulation via the ERK/DRP1 pathway, activated NF-κB/P65 nuclear translocation to promote fibrosis, and inhibitors of ERK, DRP1, and mtROS all alleviated pulmonary fibrosis lesions.
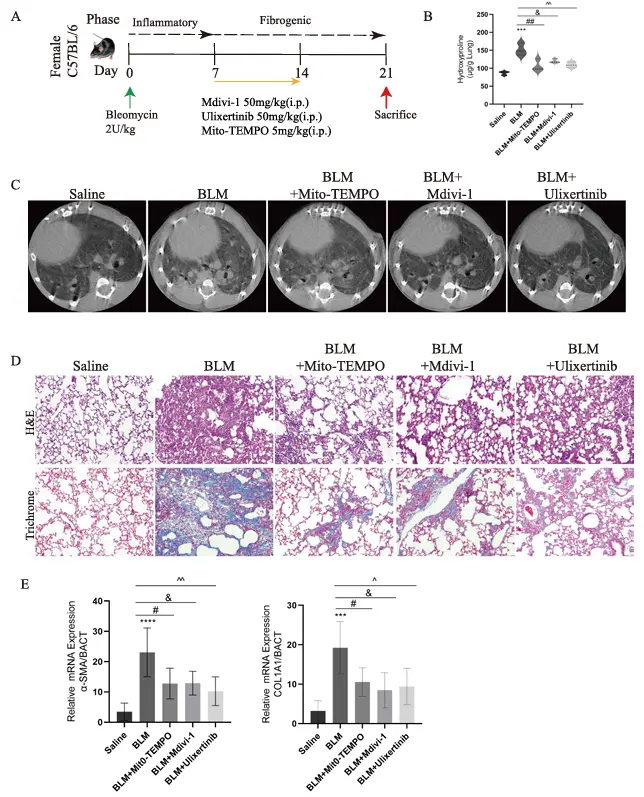
Mdivi-1 Alleviates BLM-Induced Pulmonary Fibrosis
2.3 Article Title:Mdivi-1Attenuates Sepsis-Associated Acute Lung Injury by Inhibiting M1Alveolar Macrophage Polarization and Pyroptosis

Research Overview: This study demonstrates that Mdivi-1 (a DRP1 inhibitor) reduces mitochondrial fission and Mito-ROS production by inhibiting DRP1 Ser616 phosphorylation, thereby blocking NLRP3 inflammasome activation, suppressing M1 polarization and pyroptosis in alveolar macrophages, and ultimately improving sepsis-associated acute lung injury (ALI).
The aim of this study was to investigate the mechanism by which Mdivi-1 improves LPS-induced acute lung injury (ALI) through the regulation of the DRP1/mitochondrial ROS/NLRP3 pathway. In animal experiments, an LPS-sepsis-induced ALI model was established in C57BL/6 mice, with control, LPS, and LPS+Mdivi-1 groups. In vitro experiments used RAW264.7 macrophages as a model, employing LPS stimulation combined with inhibitor or Si-DRP1 intervention. Validation was performed through histopathology, wet-to-dry ratio analysis, inflammatory cytokine assays, and molecular experiments. Results showed that Mdivi-1 significantly reduced alveolar edema and inflammatory infiltration, lowered lung injury scores and the W/D ratio, inhibited macrophage M1 polarization, reduced pro-inflammatory cytokine release, suppressed DRP1(S616) phosphorylation, scavenged mitochondrial ROS, and blocked NLRP3 inflammasome activation and pyroptosis. Si-DRP1 mimics the protective effects of Mdivi-1, confirming that Mdivi-1 exerts its anti-lung injury effects by inhibiting DRP1-mediated mitochondrial stress and the NLRP3
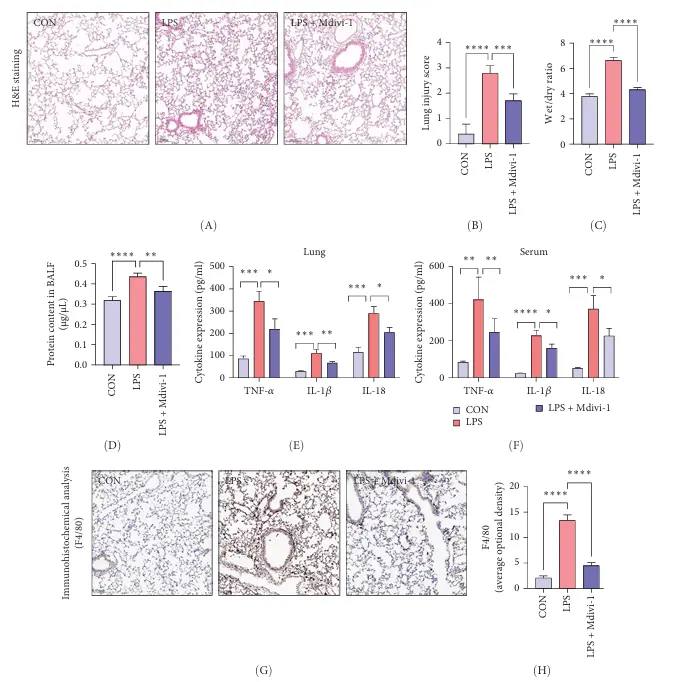
Mdivi-1 Alleviates LPS-Induced Lung Injury and Inflammation in Mice
2.4 Article Title:The oncoprotein MUC1 facilitates breast cancer progression by promoting Pink1-dependent mitophagy via ATAD3A
destabilization
Research Overview: This study focuses on the role of the MUC1 oncoprotein in breast cancer progression. It reveals for the first time that MUC1 promotes malignant progression in breast cancer by reducing ATAD3A stability and activating Pink1-dependent mitochondrial autophagy. The study elucidates the MUC1/ATAD3A/Pink1 regulatory axis and proposes that the combined inhibition of MUC1 and mitochondrial autophagy could serve as a new therapeutic strategy for breast cancer. [5]
Mdivi-1 (catalog number T1907), a specific inhibitor of mitochondrial autophagy, serves as the core tool molecule in this study: it is used to block mitochondrial autophagy, to validate that the tumor-promoting effects of MUC1 depend on mitochondrial autophagy, and as a control treatment; reversing MUC1-mediated mitochondrial protein degradation, Pink1 stabilization, cell proliferation, microvesicle formation, and tumor growth, thereby directly demonstrating that mitochondrial autophagy is a critical step in MUC1’s oncogenic activity.
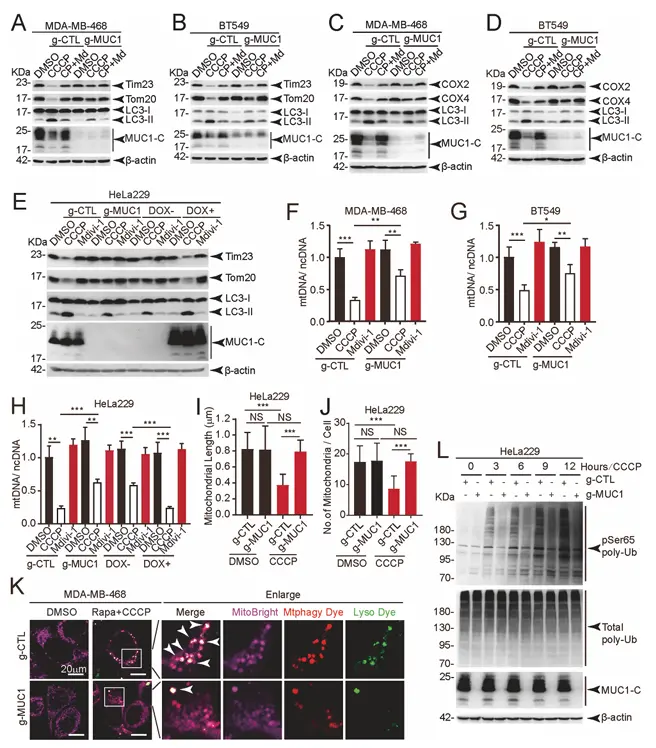
MUC1 significantly enhances CCCP-induced mitochondrial autophagy
References
[1]Jin JY,Wei XX,Zhi XL,Wang XH,Meng D.Drp1-dependent mitochondrial fission in cardiovascular disease.Acta Pharmacol Sin.2021;42(5):655-664.doi:10.1038/s41401-020-00518-y
[2]Su ZD,Li CQ,Wang HW,Zheng MM,Chen QW.Inhibition of DRP1-dependent mitochondrial fission by Mdivi-1alleviates atherosclerosis through the modulation of M1polarization.J Transl Med.2023;21(1):427.Published2023Jun30.doi:10.1186/s12967-023-04270-9
[3]Sun Z,Ji Z,Meng H,et al.Lactate facilitated mitochondrial fission-derived ROS to promote pulmonary fibrosis via ERK/DRP-1signaling.J Transl Med.2024;22(1):479.Published2024May21.doi:10.1186/s12967-024-05289-2
[4]Zhang X,Fan H,Su L,Wang Y,Chen G.Mdivi-1Attenuates Sepsis-Associated Acute Lung Injury by Inhibiting M1Alveolar Macrophage Polarization and Pyroptosis.Mediators Inflamm.2025;2025:3675276. Published2025Mar30. doi:10.1155/mi/3675276
[5] Li Q, Chu Y, Li S, et al. The oncoprotein MUC1 facilitates breast cancer progression by promoting Pink1-dependent mitophagy via ATAD3A destabilization. Cell Death Dis. 2022;13(10):899. Published 2022 Oct 26. doi:10.1038/s41419-022-05345-z
An essential round-up of science news, opinion and analysis, delivered to your inbox every weekday.
 Hello! How can I help you today?
Hello! How can I help you today? Copyright © 2015-2026 TargetMol Chemicals Inc. All Rights Reserved.