
Shopping Cart
Remove All Your shopping cart is currently empty
Your shopping cart is currently empty
Synonyms: PKU1, PKU, phenylalanine hydroxylase, PH


| Pack Size | Price | USA Stock | Global Stock | Quantity |
|---|---|---|---|---|
| 5 µg | $75 | - | In Stock | |
| 10 µg | $118 | - | In Stock | |
| 20 µg | $196 | - | In Stock | |
| 50 µg | $386 | - | In Stock | |
| 100 µg | $660 | 7-10 days | 7-10 days | |
| 200 µg | $1,120 | 7-10 days | 7-10 days | |
| 500 µg | $2,270 | 7-10 days | 7-10 days |
| Bioactivity | Activity testing is in progress. It is theoretically active, but we cannot guarantee it. If you require protein activity, we recommend choosing the eukaryotic expression version first. |
| Description | PAH (phenylalanine hydroxylase), also known as PH, belongs to the biopterin-dependent aromatic amino acid hydroxylase family. It contains 1 ACT domain, N-terminal region of PAH is thought to contain allosteric binding sites for phenylalanine and to constitute an "inhibitory" domain that regulates the activity of a catalytic domain in the C-terminal portion of the molecule. In humans, PAH is expressed both in the liver and the kidney, and there is some indication that it may be differentially regulated in these tissues. PAH catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. It is one of three members of the pterin-dependent amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin and a non-heme iron for catalysis. Defects in PAH are the cause of phenylketonuria (PKU). PKU is an autosomal recessive inborn error of phenylalanine metabolism, due to severe phenylalanine hydroxylase deficiency. It is characterized by blood concentrations of phenylalanine persistently above 1200 mumol. |
| Species | Human |
| Expression System | Baculovirus Insect Cells |
| Tag | N-His |
| Accession Number | P00439 |
| Construction | A DNA sequence encoding the human PAH (P00439) (Met 1-Lys 452) (415 Asn/Asp) was expressed, with a polyhistidine tag at the N-terminus. Predicted N terminal: Met |
| Protein Purity | > 70 % as determined by SDS-PAGE 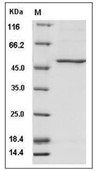 |
| Endotoxin | < 1.0 EU/μg of the protein as determined by the LAL method. |
| Formulation | Lyophilized from a solution filtered through a 0.22 μm filter, containing 20 mM Tris, 500 mM NaCl, pH 8.0, 10% glycerol. Typically, a mixture containing 5% to 8% trehalose, mannitol, and 0.01% Tween 80 is incorporated as a protective agent before lyophilization. |
| Reconstitution | Reconstituted with sterile deionized water to 0.25 mg/mL. Reconstitution conditions may vary depending on the lot. |
| Synonyms | PKU1, PKU, phenylalanine hydroxylase, PH |
| Research Background | PAH (phenylalanine hydroxylase), also known as PH, belongs to the biopterin-dependent aromatic amino acid hydroxylase family. It contains 1 ACT domain, N-terminal region of PAH is thought to contain allosteric binding sites for phenylalanine and to constitute an "inhibitory" domain that regulates the activity of a catalytic domain in the C-terminal portion of the molecule. In humans, PAH is expressed both in the liver and the kidney, and there is some indication that it may be differentially regulated in these tissues. PAH catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. It is one of three members of the pterin-dependent amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin and a non-heme iron for catalysis. Defects in PAH are the cause of phenylketonuria (PKU). PKU is an autosomal recessive inborn error of phenylalanine metabolism, due to severe phenylalanine hydroxylase deficiency. It is characterized by blood concentrations of phenylalanine persistently above 1200 mumol. |
| Molecular Weight | 54 kDa (predicted); 50 kDa (reducing conditions) |
| Shipping | In general, lyophilized powders are shipped with blue ice, while solutions are shipped with dry ice. |
| Storage | It is recommended to store recombinant proteins at -20°C to -80°C for future use. Lyophilized powders can be stably stored for over 12 months, while liquid products can be stored for 6-12 months at -80°C. For reconstituted protein solutions, the solution can be stored at -20°C to -80°C for at least 3 months. Please avoid multiple freeze-thaw cycles and store products in aliquots. |
| Size | Quantity | Unit Price | Amount | Operation |
|---|
 Hello! How can I help you today?
Hello! How can I help you today? Copyright © 2015-2026 TargetMol Chemicals Inc. All Rights Reserved.
