Peptides
Monoclonal Antibodies
Dye Reagents
PROTAC
Virtual Screening
TargetMol Kits
Cell Counting Kit-8 (CCK-8)
Inhibitor Cocktails
Natural Products
Phenols
Alkaloids
Flavonoids
Inhibitors
Angiogenesis
Apoptosis
Autophagy
Cell Cycle/Checkpoint
Chromatin/Epigenetic
Cytoskeletal Signaling
DNA Damage/DNA Repair
Endocrinology/Hormones
GPCR/G Protein
Immunology/Inflammation
JAK/STAT signaling
MAPK
Membrane transporter/Ion channel
Metabolism
Microbiology/Virology
Neuroscience
NF-Κb
oxidation-reduction
PI3K/Akt/mTOR signaling
Proteases/Proteasome
Stem Cells
Tyrosine Kinase/Adaptors
Ubiquitination
PROTAC
Others
Compound Libraries
Focused Bioactive Libraries
General Bioactive Libraries
Approved / Repurposing
Disease Focused
Target / Pathway Focused
Characteristic Bioactive Libraries
Natural Product Libraries
Natural Product Library for HTS
Characteristic Natural Product Libraries
Natural Product Derivatives Libraries
Natural Product Library for CADD
Drug-like Compound Libraries
Fragment Libraries
Custom Compound Library
Signaling Pathways
Angiogenesis
Apoptosis
Autophagy
Cell Cycle/Checkpoint
Chromatin/Epigenetic
Cytoskeletal Signaling
DNA Damage/DNA Repair
Endocrinology/Hormones
GPCR/G Protein
Immunology/Inflammation
JAK/STAT signaling
MAPK
Membrane transporter/Ion channel
Focused Bioactive Libraries
General Bioactive Libraries
Approved / Repurposing
Disease Focused
Target / Pathway Focused
Characteristic Bioactive Libraries

Cytokines and Growth Factors
• Cytokines
• Growth Factors
Receptor Proteins
• Fc Receptors
• Cytokines and Growth Factor Receptors
CD Proteins
• T Cell CD Proteins
• B Cell CD Proteins
Immune Checkpoint Proteins
• Co-inhibitory Immune Checkpoint Proteins
• Co-stimulatory Immune Checkpoint Proteins

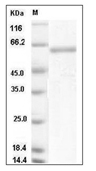
 References and Literature
References and Literature Calculator
Calculator