
Shopping Cart
Remove All Your shopping cart is currently empty
Your shopping cart is currently empty
Synonyms: SIDS, MPS2, iduronate 2-sulfatase, IDS


| Pack Size | Price | USA Stock | Global Stock | Quantity |
|---|---|---|---|---|
| 5 µg | $75 | - | In Stock | |
| 10 µg | $118 | - | In Stock | |
| 20 µg | $196 | - | In Stock | |
| 50 µg | $386 | - | In Stock | |
| 100 µg | $660 | 7-10 days | 7-10 days | |
| 200 µg | $1,120 | 7-10 days | 7-10 days | |
| 500 µg | $2,270 | 7-10 days | 7-10 days |
| Bioactivity | Measured by its ability to hydrolyze the substrate 4-Nitrocatechol Sulfate (PNCS). The specific activity is > 1.0 pmoles/min/μg. |
| Description | Iduronate 2-Sulfatase, also known as IDS, is a member of the highly conserved sulfatase family of enzymes that catalyze the hydrolysis of O- and N-sulfate esters from a variety of substrates. The human Iduronate 2-Sulfatase/IDS consists of a signal peptide, a propeptide, and a mature chain that may be further processed into two chains. Among the identified 18 human sulfatases, Iduronate 2-Sulfatase/IDS is required for the lysosomal degradation of the glycosaminoglycans (GAG), heparan sulfate, and dermatan sulfate. Multiple mutations in this X-chromosome localized gene result in Iduronate 2-Sulfatase/IDS enzymatic deficiency and lead to the sex-linked Mucopolysaccharidosis Type II (MPS II ), also known as Hunter Syndrome characterized by the lysosomal accumulation of the GAG and their excretion in urine. MPS II has a wide spectrum of clinical manifestations ranging from mild to severe due to the level of Iduronate 2-Sulfatase/IDS enzyme. Retroviral-mediated Iduronate 2-Sulfatase/IDS gene transfer into lymphoid cells would be a promising gene therapeutic strategy. |
| Species | Human |
| Expression System | HEK293 Cells |
| Tag | C-His |
| Accession Number | P22304-1 |
| Construction | A DNA sequence encoding human IDS precursor (NP_000193.1) (Met 1-Pro 550) was expressed with a C-terminal polyhistidine tag. Predicted N terminal: Ser 26 |
| Protein Purity | > 87 % as determined by SDS-PAGE 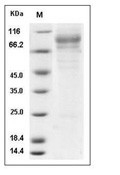 |
| Endotoxin | < 1.0 EU/μg of the protein as determined by the LAL method. |
| Formulation | Lyophilized from a solution filtered through a 0.22 μm filter, containing PBS, pH 7.4. Typically, a mixture containing 5% to 8% trehalose, mannitol, and 0.01% Tween 80 is incorporated as a protective agent before lyophilization. |
| Reconstitution | Reconstituted with sterile deionized water to 0.25 mg/mL. Reconstitution conditions may vary depending on the lot. |
| Synonyms | SIDS, MPS2, iduronate 2-sulfatase, IDS |
| Research Background | Iduronate 2-Sulfatase, also known as IDS, is a member of the highly conserved sulfatase family of enzymes that catalyze the hydrolysis of O- and N-sulfate esters from a variety of substrates. The human Iduronate 2-Sulfatase/IDS consists of a signal peptide, a propeptide, and a mature chain that may be further processed into two chains. Among the identified 18 human sulfatases, Iduronate 2-Sulfatase/IDS is required for the lysosomal degradation of the glycosaminoglycans (GAG), heparan sulfate, and dermatan sulfate. Multiple mutations in this X-chromosome localized gene result in Iduronate 2-Sulfatase/IDS enzymatic deficiency and lead to the sex-linked Mucopolysaccharidosis Type II (MPS II ), also known as Hunter Syndrome characterized by the lysosomal accumulation of the GAG and their excretion in urine. MPS II has a wide spectrum of clinical manifestations ranging from mild to severe due to the level of Iduronate 2-Sulfatase/IDS enzyme. Retroviral-mediated Iduronate 2-Sulfatase/IDS gene transfer into lymphoid cells would be a promising gene therapeutic strategy. |
| Molecular Weight | 61 kDa (predicted); 85-95 kDa (reducing condition, due to glycosylation) |
| Shipping | In general, lyophilized powders are shipped with blue ice, while solutions are shipped with dry ice. |
| Storage | It is recommended to store recombinant proteins at -20°C to -80°C for future use. Lyophilized powders can be stably stored for over 12 months, while liquid products can be stored for 6-12 months at -80°C. For reconstituted protein solutions, the solution can be stored at -20°C to -80°C for at least 3 months. Please avoid multiple freeze-thaw cycles and store products in aliquots. |
| Size | Quantity | Unit Price | Amount | Operation |
|---|
 Hello! How can I help you today?
Hello! How can I help you today? Copyright © 2015-2026 TargetMol Chemicals Inc. All Rights Reserved.
