
Shopping Cart
- Remove All
 Your shopping cart is currently empty
Your shopping cart is currently empty
APOL1, also known as apolipoprotein L1, is a minor apoprotein component of HDL (High-density lipoprotein) or 'good cholesterol' which is synthesized in the liver and also in many other tissues, including pancreas, kidney, and brain. APOL1 belongs to the apolipoprotein L family. It may play a role in lipid exchange and transport throughout the body. It may also participate in reverse cholesterol transport from peripheral cells to the liver. Defects in APOL1 are the cause of focal segmental glomerulosclerosis type 4 (FSGS4). It is a renal pathology defined by the presence of segmental sclerosis in glomeruli and resulting in proteinuria, reduced glomerular filtration rate and edema. Renal insufficiency often progresses to end-stage renal disease, a highly morbid state requiring either dialysis therapy or kidney transplantation.

| Pack Size | Price | Availability | Quantity |
|---|---|---|---|
| 50 μg | $386 | In Stock | |
| 100 μg | $660 | 7-10 days | |
| 200 μg | $1,120 | 7-10 days | |
| 500 μg | $2,270 | 7-10 days |
| Biological Activity | Activity testing is in progress. It is theoretically active, but we cannot guarantee it. If you require protein activity, we recommend choosing the eukaryotic expression version first. |
| Description | APOL1, also known as apolipoprotein L1, is a minor apoprotein component of HDL (High-density lipoprotein) or 'good cholesterol' which is synthesized in the liver and also in many other tissues, including pancreas, kidney, and brain. APOL1 belongs to the apolipoprotein L family. It may play a role in lipid exchange and transport throughout the body. It may also participate in reverse cholesterol transport from peripheral cells to the liver. Defects in APOL1 are the cause of focal segmental glomerulosclerosis type 4 (FSGS4). It is a renal pathology defined by the presence of segmental sclerosis in glomeruli and resulting in proteinuria, reduced glomerular filtration rate and edema. Renal insufficiency often progresses to end-stage renal disease, a highly morbid state requiring either dialysis therapy or kidney transplantation. |
| Species | Human |
| Expression System | Baculovirus Insect Cells |
| Tag | C-His |
| Accession Number | Q2KHQ6 |
| Synonyms | FSGS4,apolipoprotein L1,APOL-I,APOL1,APO-L,APOL |
| Construction | A DNA sequence encoding the human APOL1 (Q2KHQ6) (Met1-Leu398) was expressed with a C-terminal polyhistidine tag. Predicted N terminal: Glu 28 |
| Protein Purity | ≥ 80 % as determined by SDS-PAGE. ≥ 90 % as determined by SEC-HPLC. 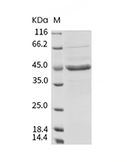 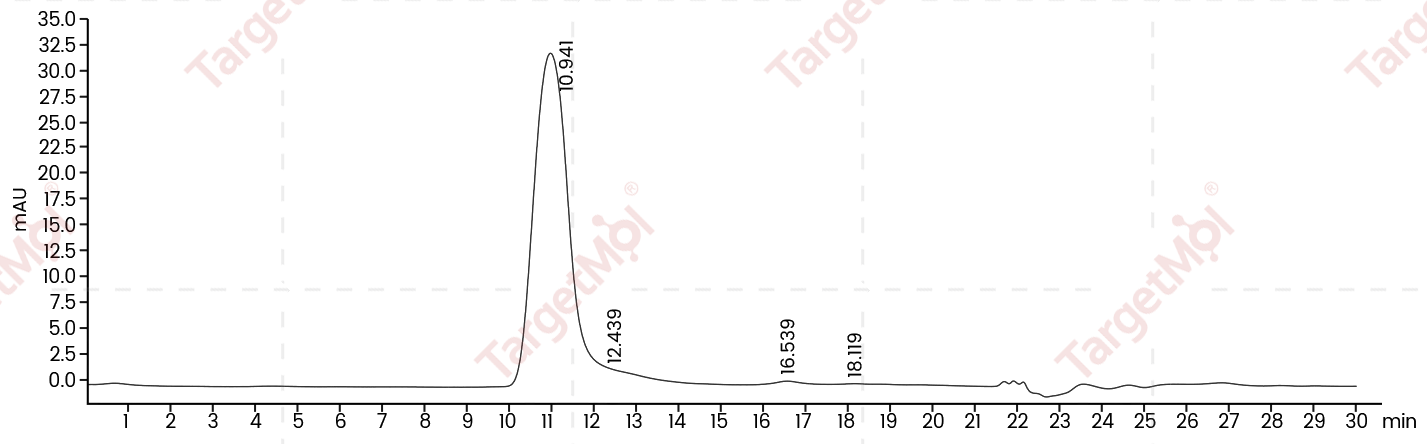 |
| Molecular Weight | 42.53 kDa (predicted); 44 kDa (reducing condition, due to glycosylation) |
| Endotoxin | < 1.0 EU/μg of the protein as determined by the LAL method. |
| Formulation | Lyophilized from a solution filtered through a 0.22 μm filter, containing 20 mM Tris, 300 mM NaCl, 10% glycerol, 0.5 mM TCEP, pH 7.5. Typically, a mixture containing 5% to 8% trehalose, mannitol, and 0.01% Tween 80 is incorporated as a protective agent before lyophilization. |
| Reconstitution | A Certificate of Analysis (CoA) containing reconstitution instructions is included with the products. Please refer to the CoA for detailed information. |
| Stability & Storage | It is recommended to store recombinant proteins at -20°C to -80°C for future use. Lyophilized powders can be stably stored for over 12 months, while liquid products can be stored for 6-12 months at -80°C. For reconstituted protein solutions, the solution can be stored at -20°C to -80°C for at least 3 months. Please avoid multiple freeze-thaw cycles and store products in aliquots. |
| Shipping | In general, Lyophilized powders are shipping with blue ice. |
| Research Background | APOL1, also known as apolipoprotein L1, is a minor apoprotein component of HDL (High-density lipoprotein) or 'good cholesterol' which is synthesized in the liver and also in many other tissues, including pancreas, kidney, and brain. APOL1 belongs to the apolipoprotein L family. It may play a role in lipid exchange and transport throughout the body. It may also participate in reverse cholesterol transport from peripheral cells to the liver. Defects in APOL1 are the cause of focal segmental glomerulosclerosis type 4 (FSGS4). It is a renal pathology defined by the presence of segmental sclerosis in glomeruli and resulting in proteinuria, reduced glomerular filtration rate and edema. Renal insufficiency often progresses to end-stage renal disease, a highly morbid state requiring either dialysis therapy or kidney transplantation. |
 Hello! How can I help you today?
Hello! How can I help you today? 
Copyright © 2015-2025 TargetMol Chemicals Inc. All Rights Reserved.